





Outre la technologie, la synthèse des glycosides a toujours suscité l'intérêt de la science, car il s'agit d'une réaction très courante dans la nature. Des articles récents de Schmidt, Toshima et Tatsuta, ainsi que de nombreuses références qui y sont citées, ont commenté un large éventail de possibilités de synthèse.
Lors de la synthèse des glycosides, un composé multisucré est associé à des nucléophiles, tels que des alcools, des glucides ou des protéines. Si une réaction sélective avec l'un des groupes hydroxyles d'un glucide est requise, toutes les autres fonctions doivent être protégées dès la première étape. En principe, les procédés enzymatiques ou microbiens, grâce à leur sélectivité, peuvent remplacer les étapes complexes de protection et de déprotection chimiques pour extraire sélectivement les glycosides dans certaines régions. Cependant, en raison de la longue histoire des alkylglycosides, l'utilisation d'enzymes dans la synthèse des glycosides n'a pas été largement étudiée et appliquée.
En raison de la capacité des systèmes enzymatiques appropriés et des coûts de production élevés, la synthèse enzymatique des alkyl polyglycosides n'est pas prête à être mise à niveau au niveau industriel et les méthodes chimiques sont préférées.
En 1870, M.A. Colley a rapporté la synthèse de « l'acétochlorhydrose » (1, figure 2) par réaction du dextrose (glucose) avec le chlorure d'acétyle, ce qui a finalement conduit à l'histoire des voies de synthèse des glycosides.
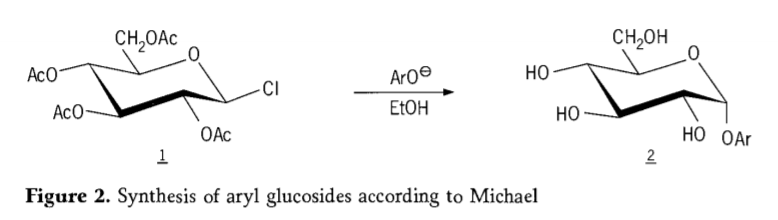
Les halogénures de tétra-0-acétyl-glucopyranosyle (acétohaloglucoses) se sont révélés plus tard utiles comme intermédiaires pour la synthèse stéréosélective de glucosides d'alkyle purs. En 1879, Arthur Michael réussit à préparer des glycosides d'aryle cristallisables à partir des intermédiaires de Colley et de phénolates (Aro-, figure 2).
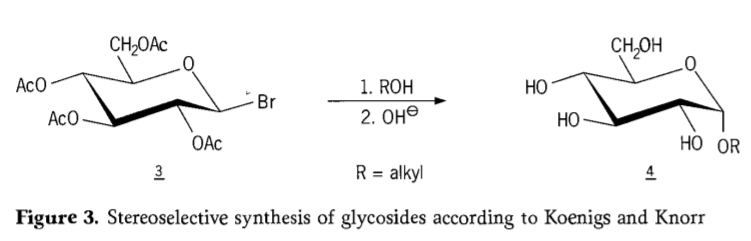
En 1901, la synthèse de Michael aboutit à une large gamme de glucides et d'aglycones hydroxylés, lorsque W. Koenigs et E. Knorr introduisirent leur procédé amélioré de glycosidation stéréosélective (figure 3). La réaction implique une substitution SN2 au niveau du carbone anomérique et se déroule de manière stéréosélective avec inversion de configuration, produisant par exemple l'α-glucoside 4 à partir de l'anomère β de l'intermédiaire acéobromoglucose 3. La synthèse de Koenigs-Knorr s'effectue en présence de promoteurs argent ou mercure.
En 1893, Emil Fischer proposa une approche fondamentalement différente pour la synthèse des alkylglucosides. Ce procédé, aujourd'hui connu sous le nom de « glycosidation de Fischer », consiste en une réaction acido-catalysée de glycoses avec des alcools. Toute analyse historique devrait néanmoins inclure la première tentative d'A. Gautier, en 1874, de conversion du dextrose par l'éthanol anhydre en présence d'acide chlorhydrique. Suite à une analyse élémentaire erronée, Gautier crut obtenir un « diglucose ». Fischer démontra plus tard que le « diglucose » de Gautier était en fait principalement de l'éthylglucoside (figure 4).
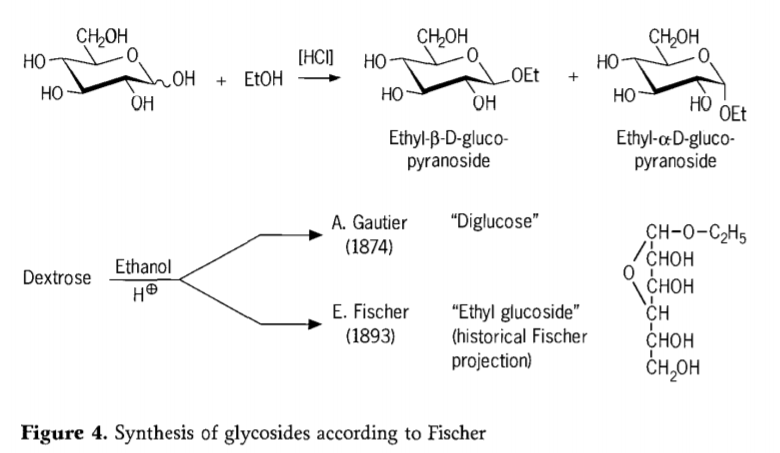
Fischer a correctement défini la structure de l'éthyl glucoside, comme le montre la formule furanosidique historique proposée. En fait, les produits de glycosidation de Fischer sont des mélanges complexes, principalement à l'équilibre, d'anomères α/β et d'isomères pyranoside/furanoside, qui comprennent également des oligomères glycosides liés aléatoirement.
Par conséquent, il est difficile d'isoler les espèces moléculaires individuelles des mélanges réactionnels de Fischer, ce qui a constitué un sérieux problème par le passé. Après quelques améliorations de cette méthode de synthèse, Fischer a ensuite adopté la synthèse de Koenigs-Knorr pour ses recherches. Grâce à ce procédé, E. Fischer et B. Helferich ont été les premiers à rapporter la synthèse d'un glucoside d'alkyle à longue chaîne présentant des propriétés tensioactives en 1911.
Dès 1893, Fischer avait correctement identifié les propriétés essentielles des alkylglycosides, telles que leur grande stabilité à l'oxydation et à l'hydrolyse, notamment en milieu fortement alcalin. Ces deux caractéristiques sont précieuses pour les alkylpolyglycosides dans les applications tensioactives.
Les recherches sur la réaction de glycosidation sont toujours en cours et plusieurs voies intéressantes pour la synthèse des glycosides ont été récemment développées. Certaines des procédures de synthèse des glycosides sont résumées dans la figure 5.
En général, les processus de glycosidation chimique peuvent être divisés en processus conduisant à des équilibres oligomères complexes dans l'échange de glycosyle catalysé par un acide.
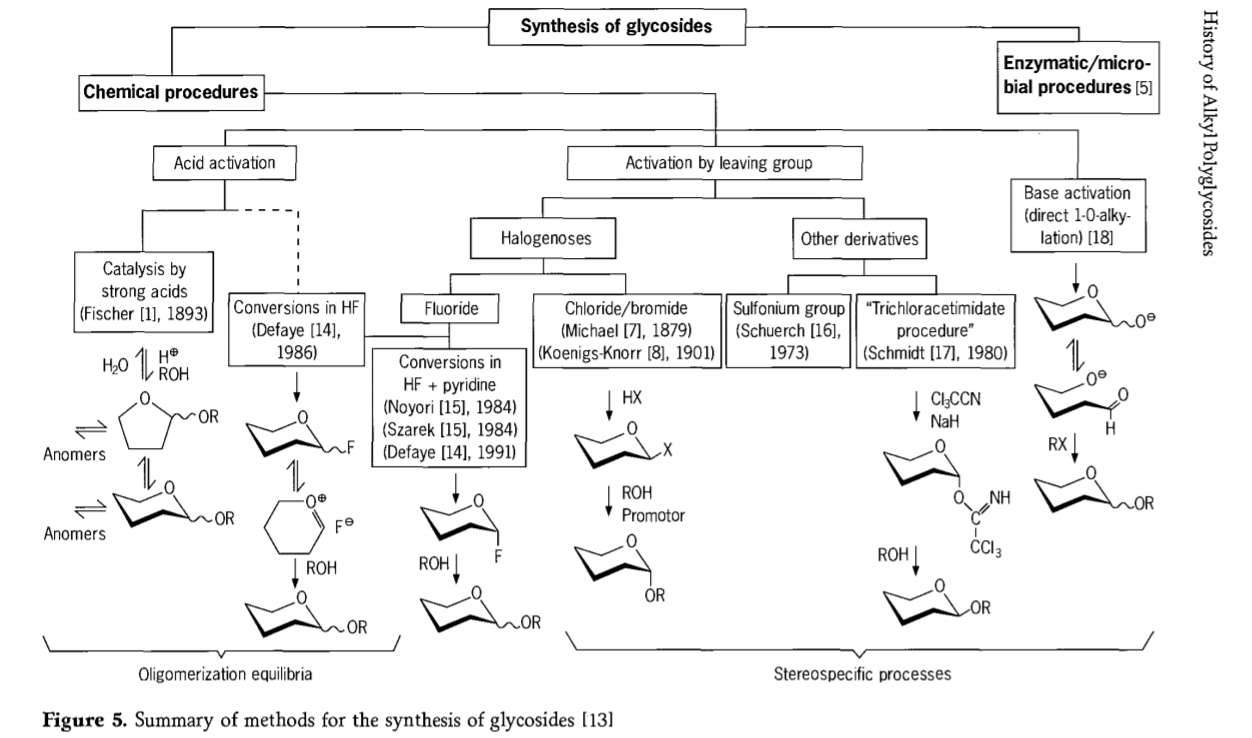
Réactions sur des substrats glucidiques correctement activés (réactions glycosidiques de Fischer et réactions au fluorure d'hydrogène (HF) avec des molécules glucidiques non protégées) et réactions de substitution cinétiquement contrôlées, irréversibles et principalement stéréotaxiques. Un deuxième type de procédure peut conduire à la formation d'espèces individuelles plutôt que de mélanges complexes de réactions, notamment lorsqu'il est combiné à des techniques de conservation des groupes. Les glucides peuvent laisser des groupes sur le carbone ectopique, tels que des atomes d'halogène, des sulfonyles ou des groupes trichloroacétimidate, ou être activés par des bases avant leur conversion en esters triflates.
Dans le cas particulier des glycosidations dans le fluorure d'hydrogène ou dans des mélanges de fluorure d'hydrogène et de pyridine (poly[fluorure d'hydrogène] de pyridinium), des fluorures de glycosyle se forment in situ et se transforment facilement en glycosides, par exemple avec des alcools. Le fluorure d'hydrogène s'est révélé être un milieu réactionnel fortement activateur et non dégradant ; une autocondensation à l'équilibre (oligomérisation) est observée, similaire au processus de Fischer, bien que le mécanisme réactionnel soit probablement différent.
Les glycosides d'alkyle chimiquement purs ne conviennent qu'à des applications très spécifiques. Par exemple, ils ont été utilisés avec succès en recherche biochimique pour la cristallisation de protéines membranaires, comme la cristallisation tridimensionnelle de la porine et de la bactériorhodopsine en présence d'octyl β-D-glucopyranoside (d'autres expériences basées sur ces travaux ont valu à Deisenhofer, Huber et Michel le prix Nobel de chimie en 1988).
Au cours du développement des alkylpolyglycosides, des méthodes stéréosélectives ont été utilisées en laboratoire pour synthétiser diverses substances modèles et étudier leurs propriétés physicochimiques. En raison de leur complexité, de l'instabilité des intermédiaires et de la quantité et de la nature critique des déchets de procédé, les synthèses de type Koenigs-Knorr et autres techniques de groupes protecteurs poseraient d'importants problèmes techniques et économiques. Les procédés de type Fischer sont comparativement moins complexes et plus faciles à mettre en œuvre à l'échelle industrielle et constituent donc la méthode privilégiée pour la production d'alkylpolyglycosides à grande échelle.
Date de publication : 12 septembre 2020